


Optical experiments on single molecules at room temperature are currently affected by two major limitations: On one hand it is not possible to have arbitrarily long continuous observation periods of one and the same chromophore because of the inevitable occurrence of photoblinking and photobleaching events. For short times, on the other end, the limited number of photons per second which can be emitted by a single molecule require a certain minimum integration time to detect a signal above the omnipresent shot noise. We have developed a novel type of temperature-cycle microscopy to address both these problems.
Our method relies on rapid thermal cycling of a microscopic sample to separate the room-temperature conformational evolution of, for example, a single biomolecule from the optical probing of the chromophore(s)/label(s), which takes place under cryogenic conditions. That way we want to probe a frozen “snapshot” of the biomolecule at a temperature where the photophysical parameters of the label(s) are more favorable than at room temperature. In that case the temporal resolution is given by the time it takes to undergo the temperature-jump and is no longer limited by photon statistics. Furthermore, the controlled manipulation of temperature allows one to investigate energetic barriers between conformational sub-states.

The core components of our microscope, which are integrated in an optical cryostat, are shown in the diagram above. A high-NA microscope objective is used to both focus the optical probing laser and collect the emitted fluorescence, as part of a laser-scanning confocal microscope. The sample film is spin-coated onto an absorbing metal layer which allows us to generate a small hot spot by focusing a near-IR heating beam with an aspheric singlet lens. Additional optics outside of the cryostat make it possible to control the position of the heating spot on the sample.
We use fluorescence anisotropy of rhodamine 6G in glycerol to visualize and quantify the effect of our heating laser on the sample film: An ensemble of randomly oriented molecules is excited with linearly polarized laser light and we record the fluorescence intensity which is emitted with parallel and perpendicular polarization in two separate detection channels. As long as the molecules are immobilized in a frozen matrix, most of the fluorescence will be emitted with a polarization parallel to the excitation polarization. At higher temperatures, when the viscosity of the matrix is reduced and the chromophores start to rotate significantly during the fluorescence lifetime (a few nanoseconds), a larger part of the fluorescence will be depolarized which results in a lower fluorescence anisotropy. We chose glycerol because its viscosity depends very strongly on temperature, which results in the following calibration curve (anisotropy versus temperature):
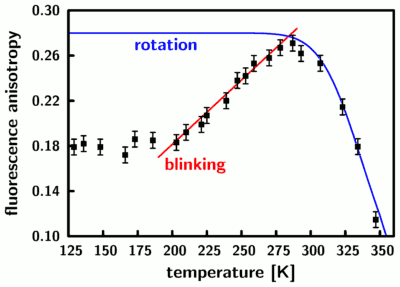
We find that the measured anisotropy agrees very well with what can be expected from known material properties of glycerol (blue curve) for temperatures above 280 Kelvin. Below that temperature we find a reduction of the anisotropy due to saturation effects which are caused by the photoblinking of rhodamine 6G. The above calibration curve allows us to image the effect of our focused heating laser on the sample and thus measure local temperatures with microscopic resolution:
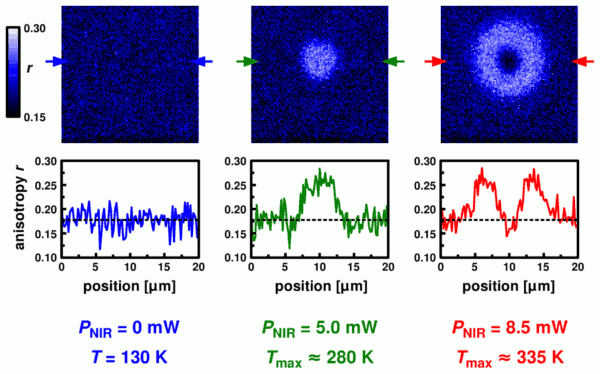
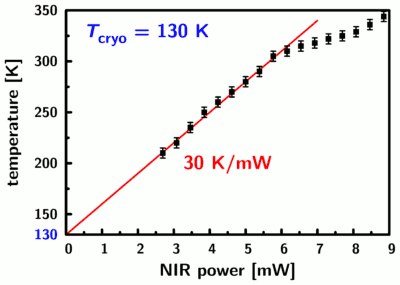
We find a uniform anisotropy in the image to the right which reflects the situation at 130 Kelvin without local heating. With increasing near-IR heating power we first see an increase of the anisotropy due to the temperature-dependent saturation behavior (center image and line profile) and then the anisotropy decreases again due to the accelerated rotation of the chromophores (left hand side). The dimensions of the heating spot are on the order of 5 microns (full-width at half-maximum). The response to the NIR heating beam is linear up to a temperature in the center of the hot spot of about 300 Kelvin; at higher temperatures the response of the system is changed due to increasing contributions from convection:
As pointed out in the beginning, the heating and cooling times are an important parameter for our technique since they determine our temporal resolution. We conducted repeated heating/cooling cycles by modulating the heating laser at a frequency of 2.5 kHz and recording the arrival time of each detected photon relative to the start of the current cycle. Taking the data from thousands of cycles, we can then determine the changes in temperature (from the anisotropy) with a resolution of 1 microsecond:
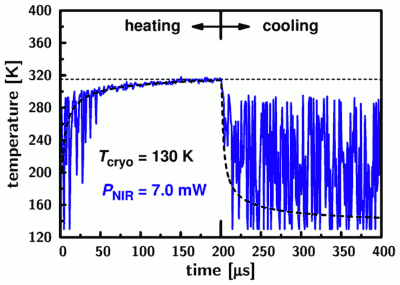
The data (blue line) is noisy for temperatures below 280 Kelvin due to the decreased sensitivity of the anisotropy in that temperature range, but it nevertheless demonstrates directly how fast we reach room temperature when starting from 130 Kelvin. A quantitative analysis using a simple heat diffusion model (thick dashed line) yields a transition half-time of only 3.5 microseconds. This is 2-3 orders of magnitude shorter that the temporal resolution limit due to photon counting statistics which is encountered in conventional single-molecule experiments.
References
- R. Zondervan, F. Kulzer, H. van der Meer, J.A.J.M. Disselhorst, M. Orrit
“Laser-driven microsecond temperature cycles analyzed by fluorescence polarization microscopy”
Biophys. J. 90 (2006) 2958-2969 (a BioFAST pre-print is also available)